...
for support, contact us (hpc-support@lists.cnaf.infn.it).
- SLURM architecture
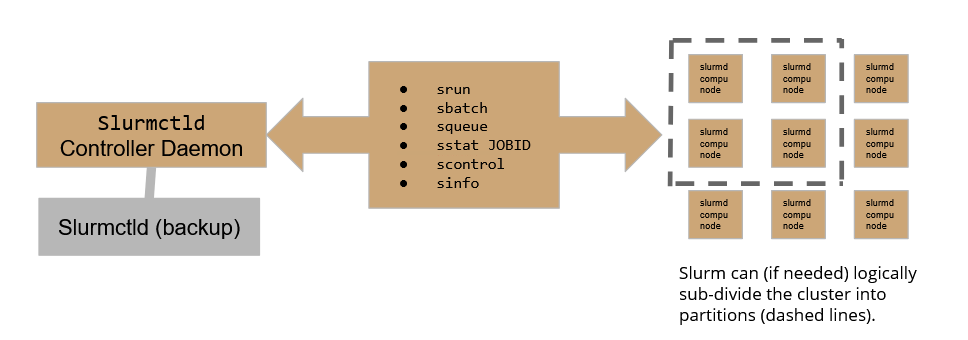
the Slurmctld daemon runs on ui-hpc, which therefore plays the role of submit point of the cluster. A backup daemon is configured to assure the continuity of service.
On our HPC cluster, there are currently 4 active partitions:
- slurmHPC_int: MaxTime allowed for computation = 2h
- slurmHPC_inf: MaxTime allowed for computation = 79h
- slurmHPC_short: short MaxTime allowed for computation = 79h
- slurmHPC_gpu: MaxTime allowed for computation = 33h
Please be aware that exceeding the MaxTime enforced will result in the job being held.
if not requested differently at submit time, jobs will be submitted to the _int partition. Users can choose freely what partition to use by configuring properly the batch submit file (see below).
- Check the cluster status with SLURM
...
A Slurm batch job can be configured quite extensively, so shedding some light on the most common sbatch options may help us configuring jobs properly.
#SBATCH --partition=<NAME>
This instruction allows the user to specify on which queue (partition in Slurm jargon) the batch job must land. The option is case-sensitive.
#SBATCH --job-name=<NAME>
This instruction assigns a “human-friendly” name to the job, making it easier for the user to recognize among other jobs.
#SBATCH --output=<FILE>
This instructions allows to redirect any output to a file (dynamically created at run-time)
#SBATCH --nodelist=<NODES>
This instruction forces Slurm to use a specific subset of nodes for the batch job. For example: if we have a cluster of five nodes: node[1:5] and we specify --nodelist=node[1-2], our job will only use these two nodes.
#SBATCH --nodes=<INT> <INT>
This instructions tells slurm to run over <INT> random nodes belonging to the partition.
N.B. Slurm chooses the best <INT> nodes evaluating current payload, so the choice is not entirely random. If we want specific nodes to be used, the correct option is the aforementioned --nodelist
#SBATCH --ntasks=<INT>
This command tells Slurm to use <INT> CPUS to perform the job. The CPU load gets distributed to optimize the efficiency and the computational burden on nodes
#SBATCH --ntasks-per-node=<INT>
This command is quite different from the former one: in this case Slurm forces the adoption of <INT> CPUS per node. Suppose you chose 2 nodes for your computation, writing --ntasks-per-node=4, you will force the job to use 4 CPUS on the first node as well as 4 CPUS on the second one.
#SBATCH --time=<TIME>
This command sets an upper time limit for the job to be considered running. When this limit is exceeded, the job will be automatically held.
#SBATCH --mem=<INT>
This option sets an upper limit for memory usage on every compute node in the cluster. It must be coherent with node hardware capabilities in order to avoid failures
...
In the following we present some advanced SBATCH options. These ones will allow the user to set up constraints and use specific computing hardware peripherals, such as GPUs.
#SBATCH --constraint=<...>
This command sets up hardware constraints. This allows the user to specify over which hardware the job should land or should leverage. Some examples may be: --constraint=IB (use forcibly Infini-Band nodes) or --constraint=<CPUTYPE> (use forcibly CPUTYPE CPUs)
#SBATCH --gres=<GRES>:<#GRES>
This command allows (if gres are configured) leveraging general computing resources. Typical use-case: GPUs. In that case, the command looks like:
--gres=gpu:<INT> where <INT> is the number of GPUs we want to use.
#SBATCH --mem-per-cpu=<INT>
This command sets a memory limit CPU-wise.
...
In the following, some examples of submit files to help the user get comfortable with Slurm. See them as a controlled playground to test some of the features of Slurm
Simple batch submit
#!/bin/bash
#
#SBATCH --job-name=tasks1
#SBATCH --output=tasks1.txt
#SBATCH --nodelist=hpc-200-06-[17-18]
#SBATCH --ntasks-per-node=8
#SBATCH --time=5:00
#SBATCH --mem-per-cpu=100
srun hostname -s
the output should be something like (hostnames and formatting may change):
...
As we can see the execution involved 8 CPUs only and the payload was organized to minimize the burden over the nodes.
Simple MPI submit
#!/bin/bash
#
#SBATCH --job-name=test_mpi_picalc
#SBATCH --output=res_picalc.txt
#SBATCH --nodelist=... #or use --nodes=...
#SBATCH --ntasks=8
#SBATCH --time=5:00
#SBATCH --mem-per-cpu=1000
srun picalc.mpi
If the .mpi file is not available on compute nodes, which will be the most frequent scenario if you save your files in a custom path, computation is going to fail. That happens because Slurm does not take autonomously the responsibility of transfering files over compute nodes.
...
build the srun command as follows:
srun --bcast=~/picalc.mpi picalc.mpi
Where the --bcast option copies the executable to every node by specifying the destination path. In this case, we decided to copy the executable into the home folder keeping the original name as-is.
...