If your use case has needs related to parallel computing, i.e. multinode-MPI or needs special computing resources such as Intel Manycore or NVIDIA GPUs, probably you need to access the CNAF HPC cluster, which is a small cluster (about 100TFLOPS) but with special HW available [9].
This cluster is accessible via the LSF batch system for which you can find instructions here [10] and Slurm Workload Manager which will be introduced hereafter. Please keep in mind that the LSF solution is currently in its EOL phase and will be fully substituted by Slurm Workload Manager in the near future.
In the following, few details on the account request and first access are given.
Account Request
If a user already has an hpc account, it can skip this paragraph. Otherwise, some preliminary steps occur.
Download and fill in the access authorization form. If help is needed, please contact us at hpc-support@lists.cnaf.infn.it.
Send the form via mail to sysop@cnaf.infn.it and to user-support@lists.cnaf.infn.it
Once the account creation is completed, you will receive a confirmation email at the specified address. In that occasion, you will receive the credentials you need to access the cluster as well, i.e. username and temporary password.
Access
First of all, it is necessary SSH into bastion.cnaf.infn.it with the own credentials.
Thereafter, access the HPC cluster logging into ui-hpc.cr.cnaf.infn.it from bastion. You have to use the same credentials you just used to log into bastion.
Your home directory will be: /home/HPC/<your_username>. The home folder is shared among all the cluster nodes.
No quotas are currently enforced on the home directories and about only 4TB are available in the /home partition. In case you need more disk space for data and checkpointing, every user can access the following directory:
/storage/gpfs_maestro/hpc/user/<your_username>/
which is on a shared gpfs storage. Please, do not leave huge unused files in both home directories and gpfs storage areas. Quotas will be however enforced in the near future.
For support, contact us (hpc-support@lists.cnaf.infn.it).
SLURM architecture
Slurm workload manager relies on the following scheme:
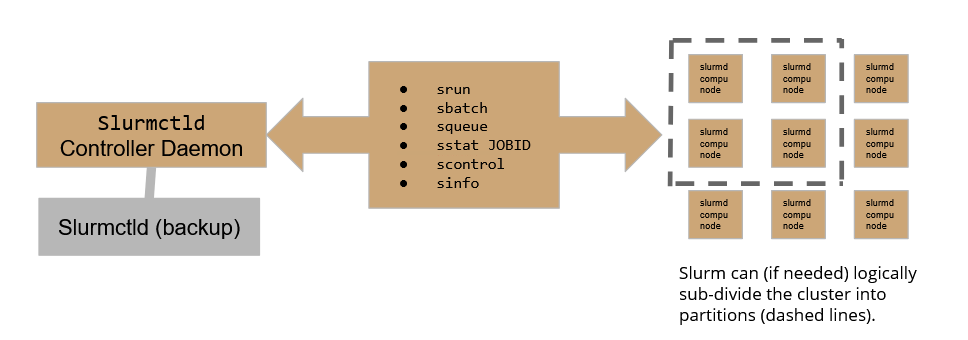
Where the Slurmctld daemon plays the role of the controller, allowing the user to submit and follow the execution of a job, while Slurmd daemons are the active part in the execution of jobs over the cluster. To assure high availability, A backup controller daemon has been configured to assure the continuity of service.
On our HPC cluster, there are currently 4 active partitions:
- slurmHPC_int MaxTime allowed for computation = 79h
- slurmHPC_inf MaxTime allowed for computation = 79h
- slurmHPC_short MaxTime allowed for computation = 79h
- slurm_GPU MaxTime allowed for computation = 33h
- slurm_hpc_gpuV100 MaxTime allowed for computation = 33h
Please be aware that exceeding the MaxTime enforced will result in the job being held.
If not requested differently at submit time, jobs will be submitted to the _int partition. Users can choose freely what partition to use by configuring properly the batch submit file (see below).
Check the cluster status with SLURM
You can check the cluster status using the sinfo -N command which will print a summary table on the standard output.
The table shows 4 columns: NODELIST, NODES, PARTITION and STATE.
- NODELIST shows node names. Multiple occurrences are allowed since a node can belong to more than one partition
- NODES indicates the number of machines available.
- PARTITION which in slurm is a synonym of "queue" indicates to which partition the node belongs. If a partition name comes with an ending asterisk, it means that that partition will be considered the default one to run the job, if not otherwise specified.
- STATE indicates if the node is not running jobs ("idle"), if it is in drain state ("drain") or if it is running some jobs ("allocated").
For instance:
-bash-4.2$ sinfo -N NODELIST NODES PARTITION STATE hpc-200-06-05 1 slurmHPC_int* idle hpc-200-06-05 1 slurmHPC_short idle hpc-200-06-05 1 slurmHPC_inf idle hpc-200-06-05 1 slurm_GPU idle hpc-200-06-06 1 slurmHPC_short idle hpc-200-06-06 1 slurmHPC_int* idle hpc-200-06-06 1 slurmHPC_inf idle [...]
The structure of a basic batch job
To work with a batch, the user should build a batch submit file. Slurm accepts batch files that respect the following basic syntax:
#!/bin/bash # #SBATCH <OPTIONS> (...) srun <INSTRUCTION>
The #SBATCH directive teaches Slurm how to configure the environment for the job at matter, while srun is used to actually execute specific commands. It is worth now getting acquainted of some of the most useful and commonly used options one can leverage in a batch submit file.
In the following, the structure of batch jobs alongside with few basic examples will be discussed.
Submit basic instructions on Slurm with srun
In order to run instructions, a job has to be scheduled on Slurm: the basic srun command allows to execute very simple commands, such as one-liners on compute nodes.
The srun command can be also enriched with several useful options:
- N<int> allows to run the command on <int> nodes, for instance:
srun -N5 /bin/hostnamewill run the hostname command on 5 nodes;
- v option activates verbosity (vvv is max verbosity);
- w option activates manual node selection.
If the user needs to specify the run conditions in detail as well as running more complex jobs, the user must write down a batch submit file.
#SBATCH options
A Slurm batch job can be configured quite extensively, so shedding some light on the most common sbatch options may help us configuring jobs properly.
To have a more complete list of command options you can visit the slurm documentation. [30]
#SBATCH --partition=<NAME>
This instruction allows the user to specify on which queue (partition in Slurm jargon) the batch job must land. The option is case-sensitive.#SBATCH --job-name=<NAME>
This instruction assigns a “human-friendly” name to the job, making it easier for the user to recognize among other jobs.#SBATCH --output=<FILE>
This instructions allows to redirect any output to a file (dynamically created at run-time).#SBATCH --nodelist=<NODES>
This instruction forces Slurm to use a specific subset of nodes for the batch job. For example: if we have a cluster of five nodes:node[1:5]and we specify--nodelist=node[1-2], our job will only use these two nodes.#SBATCH --nodes=<INT>
This instructions tells slurm to run over <INT> random nodes belonging to the partition.
N.B. Slurm chooses the best <INT> nodes evaluating current payload, so the choice is not entirely random. If we want specific nodes to be used, the correct option is the aforementioned --nodelist.#SBATCH --ntasks=<INT>
This command tells Slurm to use <INT> CPUS to perform the job. The CPU load gets distributed to optimize the efficiency and the computational burden on nodes.#SBATCH --ntasks-per-node=<INT>
This command is quite different from the former one: in this case Slurm forces the adoption of <INT> CPUS per node. Suppose you chose 2 nodes for your computation, writing --ntasks-per-node=4, you will force the job to use 4 CPUS on the first node as well as 4 CPUS on the second one.#SBATCH --time=<TIME>
This command sets an upper time limit for the job to be considered running. When this limit is exceeded, the job will be automatically held.#SBATCH --mem=<INT>
This option sets an upper limit for memory usage on every compute node in the cluster. It must be coherent with node hardware capabilities in order to avoid failures
Advanced batch job configuration
In the following we present some advanced SBATCH options. These ones will allow the user to set up constraints and use specific computing hardware peripherals, such as GPUs.
#SBATCH --constraint=<...>
This command sets up hardware constraints. This allows the user to specify over which hardware the job should land or should leverage. Some examples may be:--constraint=IB(use forcibly Infini-Band nodes) or--constraint=<CPUTYPE>(use forcibly CPUTYPE CPUs).#SBATCH --gres=<GRES>:<#GRES>
This command allows (if gres are configured) leveraging general computing resources. Typical use-case: GPUs. In that case, the command looks like:--gres=gpu:<INT>where<INT>is the number of GPUs we want to use.#SBATCH --mem-per-cpu=<INT>
This command sets a memory limit CPU-wise.
N.B. if one sets this instruction, it is mandatory for it to be the only constraint on --mem in the batch job configuration.
In the following, few utilization examples are given in order to practice with these concepts.
Retrieve job information
A user can retrieve information regarding active job queues with the squeue command for a synthetic overview of Slurm job queue status.
Among the information printed with the squeue command, the user can find the job id as well as the running time and status.
In case you are running a job, you can have a detailed check on it with the command
sstat -j <jobid>
where jobid is an id given to you by Slurm once you submit the job.
Migrating from LSF
Down below are listed some frequently used LSF commands and their SLURM equivalents.
Essential commands
| LSF | SLURM | Description |
|---|---|---|
| bsub < script_file | sbatch script_file | Submit a job script to the scheduler |
| bqueues | sinfo | Show available scheduling queues |
| bjobs | squeue -u <username> | List user's pending and running jobs |
| bsub -n 1 -Is /bin/bash | srun -n 1 --pty /bin/bash | Request an interactive session |
Job specification
| LSF | SLURM | Description |
|---|---|---|
| #BSUB | #SBATCH | Scheduler directive |
| -q queue_name | --partition=queue_name or -p queue_name | Specify the scheduling queue |
| -W hh:mm:ss | --time= hh:mm:ss or -t hh:mm:ss | Set the maximum runtime limit |
| -We hh:mm:ss | --time-min= hh:mm:ss | Set an estimated runtime |
| -J job_name | --job-name= jobname | Specify a name for the job |
-o filename | --output= filename or -o filename, | Standard job output |
| -e filename | --error=filename or -e filename | Standard job error output |
| -n number-of-cores | --ntasks= number-of-cores or -n number-of-cores | Number of CPU cores |
| -m <host-group-name> | --constraint="<host-group-name>" | To select a node with a specific processor model |
Further commands and options are listed at the following link [32].
Examples
Below some examples of submit files follow to help the user get comfortable with Slurm.
See them as a controlled playground to test some of the features of Slurm.
Simple batch submit
#!/bin/bash # #SBATCH --job-name=tasks1 #SBATCH --output=tasks1.txt #SBATCH --nodelist=hpc-200-06-[17-18] #SBATCH --ntasks-per-node=8 #SBATCH --time=5:00 #SBATCH --mem-per-cpu=100 srun hostname -s
To execute this script, the command to be issued is
sbatch <executable.sh>
bash-4.2$ sbatch test_slurm.sh Submitted batch job 8501
To retrieve the information about the submitted jobs you can use the command "squeue":
bash-4.2$ squeue
JOBID PARTITION NAME USER ST TIME NODES NODELIST(REASON)
8501 slurmHPC_ test_sl apascoli R 0:01 1 hpc-200-06-05
The table shows 8 columns: JOBID, PARTITION, NAME, USER, ST, TIME, NODES and NODELIST(REASON).
- JOBID shows the id corresponding to the submitted jobs.
- PARTITION which in slurm is a synonym of "queue" indicates to which partition the node belongs.
- NAME it corresponds to the name assigned in the submit file, otherwise it will match the name of the submit file.
- USER indicates the user who submitted the job.
- ST indicates if the jobs are running ("R") or if it is pending ("PD").
- TIME shows how long the jobs have run for using the format days-hours:minutes:seconds.
- NODES indicates the number of machines running the job.
- NODELIST indicates where the job is running or the reason it is still pending.
Then, to see information about a single job use:
bash-4.2$ sstat --format=JobID,AveCPU -j 8501 JobID AveCPU ------------ ---------- 8501.0 213503982+
The option "--format" allows to customise the output based on the desired features.
For instance in the example above are shown:
- JobID
- AveCPU Average (system + user) CPU time of all tasks in job.
Many more features are listed in the slum manual. [31]
The output, from the option --output=tasks1.txt, should be something like (hostnames and formatting may change):
-bash-4.2$ cat tasks1.txt hpc-200-06-17 hpc-200-06-17 hpc-200-06-17 hpc-200-06-17 hpc-200-06-17 [...]
As we can see, the --ntasks-per-node=8 option was interpreted by slurm as “reserve 8 cpus on node 17 and 8 on node 18 and execute the job on those cpus”.
It is quite useful to see how the output would look in case of --ntasks=8 was used instead of --ntasks-per-cpu. In that case, the output should be exactly:
hpc-200-06-17 hpc-200-06-18 hpc-200-06-17 hpc-200-06-18 hpc-200-06-17 hpc-200-06-18 hpc-200-06-17 hpc-200-06-18
As we can see the execution involved 8 CPUs only and the payload was organized to minimize the burden over the nodes.
Simple MPI submit
To submit an MPI job on the cluster, the user must secure-copy the .mpi executable over the cluster.
In the following example, we prepared a C script that calculates the value of pi and we compiled it with:
module load mpi/openmpi-x86_64 mpicc example.c -o picalc.mpi
N.B: before compiling the file, you need to load the proper module as shown above. You can find the available modules by running module avail. Finally, it is possible to see the list of the loaded modules with:
-bash-4.2$ module list Currently Loaded Modulefiles: 1) mpi/openmpi-x86_64
Here's an example of a submit file:
#!/bin/bash # #SBATCH --job-name=test_mpi_picalc #SBATCH --output=res_picalc.txt #SBATCH --nodelist=... #or use --nodes=... #SBATCH --ntasks=8 #SBATCH --time=5:00 #SBATCH --mem-per-cpu=1000 srun picalc.mpi
If the .mpi file is not available on desired compute nodes, which will be the most frequent scenario if you save your files in a custom path, computation is going to fail. That happens because Slurm does not take autonomously the responsibility of transferring files over compute nodes.
On way of acting might be to secure copy the executable over the desired nodelist, which can be feasible only if 1-2 nodes are involved. Otherwise, Slurm offers a srun option which may help the user.
In this case, the srun command has to be built as follows:
srun --bcast=~/picalc.mpi picalc.mpi
Where the --bcast option copies the executable to every node by specifying the destination path. In this case, we decided to copy the executable into the home folder keeping the original name as-is.
Simple Python submit
The submission of a python job over the cluster follows the syntactic rules of previous use-cases. The only significative difference is that we have to specify the compiler/interpreter when we call the srun command.
In the following, we show how to execute a python job over 16 CPUs of a selected cluster node.
#!/bin/bash # #SBATCH --job-name=prova_python_sub #SBATCH --output=prova_python_sub.txt #SBATCH --nodelist=hpc-200-06-05 #SBATCH --ntasks=16 #SBATCH --time=5:00 #SBATCH --mem-per-cpu=100 srun python3 trial.py
Again, if secure-copying the executable over every node involved in our computation is an un-optimized operation, the user should add the --bcast option to the srun command.
Python submit with a virtual environment
With Slurm, we can also submit python jobs that leverage a virtual environment. This comes handy if the job needs to use packages that are not included in the standard python setup on the cluster.
In order to do so, the user has to:
- Create a virtual environment in Python
- Activate the environment
- Install the packages that the job needs
- Deactivate the environment
These 4 steps are required only once.
Henceforth, the user can add these lines to the submission script and use the desired virtual environment:
source PATH_TO_VENV srun python myscript.py deactivate
Additional information
Complete overview of hardware specs per node:http://wiki.infn.it/strutture/cnaf/clusterhpc/home.
Ask for support via E-mail: hpc-support@lists.cnaf.infn.it.